About Us
News

Modeling regulatory cis-elements for functional annotation of transcription-modulating genetic variants
The human genome carries the instruction manuals for gene expression regulation. Deciphering the “genetic codes” encoded in human genome through computational approaches has emerged as a central theme in our lab. Cells are the fundamental building blocks of life. Several kinds of functional cis- elements are involved in gene expression regulation of cells: more than Read more about Modeling regulatory cis-elements for functional annotation of transcription-modulating genetic variants[…]

An accurate computational method for cross-platform, multi-modal spatial omics alignment and integration.
Life is an organized arrangement of cells, but any individual cell struggles to function independently once detached from a living organism. Therefore, a comprehensive understanding of cell functionality requires considering the microenvironment and spatial location in which cells exist. The rapid development of spatial omics technology greatly facilitates the study of cell function at the Read more about An accurate computational method for cross-platform, multi-modal spatial omics alignment and integration.[…]
Mol. Biol. Evol. | Genome-wide identification of gene loss events suggests loss relics as a potential source of functional lncRNAs in humans
Through the process of transcription and translation, protein-coding genes (hereinafter referred to as genes) can guide the synthesis of proteins that are crucial to life activities, and then affect the physiological or pathological traits of living organisms. Thus, genes are the basic functional units in cells. Research in the past decades has shown that in Read more about Mol. Biol. Evol. | Genome-wide identification of gene loss events suggests loss relics as a potential source of functional lncRNAs in humans[…]

GLUE for China’s top ten bioinformatics advances of 2022
The GLUE study (Multi-omics single-cell data integration and regulatory inference with graph-linked embedding) published in Nature Biotechnology was recently awarded by Genomics, Proteomics and Bioinformatics (GPB) as China’s top ten bioinformatics advances of 2022. With rapid advances in recent years, single-cell omics technologies have become essential tools for studying cellular function and gene regulation. Read more about GLUE for China’s top ten bioinformatics advances of 2022[…]

Nat. Biotechnol. | An accurate and robust computational method for single-cell multi-omics integration and regulatory inference
Nat. Biotechnol. | An accurate and robust computational method for single-cell multi-omics integration and regulatory inference Gene transcription is a key link in the central dogma of biology. Compared to the relative static genome, the transcriptome exhibits substantial changes across different tissues, organs and developmental stages, forming crucial biological basis for the physiological and pathological Read more about Nat. Biotechnol. | An accurate and robust computational method for single-cell multi-omics integration and regulatory inference[…]
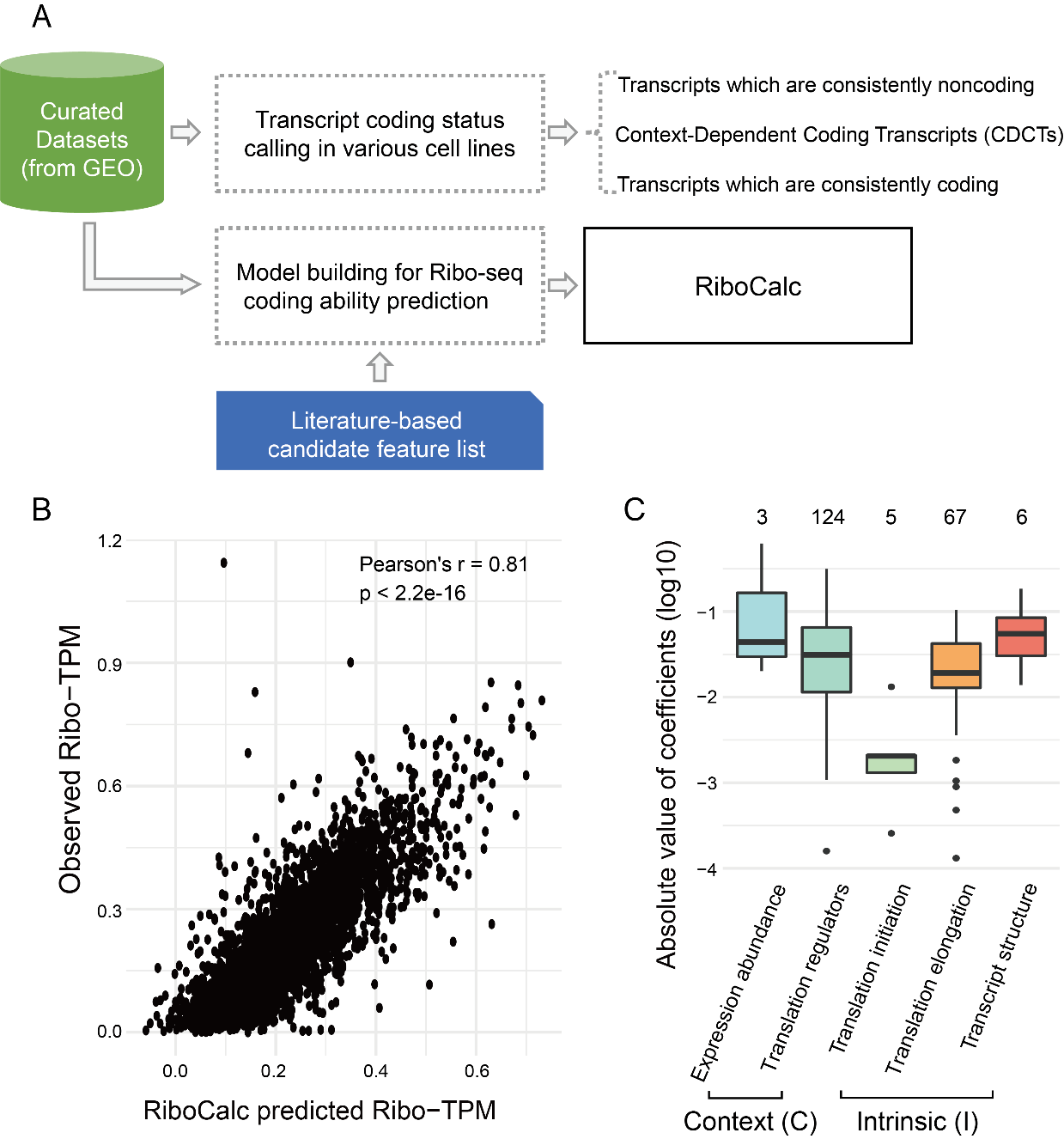
RiboCalc: Quantitative model suggests both intrinsic and contextual features contribute to the transcript coding ability determination in cells
Gene transcription and protein translation are two key steps of the “central dogma”. Cells often response to disease and environment stress via transcriptional and translational control. Meanwhile, besides coding proteins, RNAs could function as noncoding molecules. Benefitting from bioinformatics and computational genomics methods, it is possible to quantitatively de-convolute factors contributing to translational control in Read more about RiboCalc: Quantitative model suggests both intrinsic and contextual features contribute to the transcript coding ability determination in cells[…]

Welcome ShiQI Yang to join the Gao lab
ShiQi Yang is the first staff join us via the changping lab. Warmly welcome!

Welcome XianYi Lian to join the Gao lab and congratulation her on winning the Peking University Boya Postdoctoral Fellowship
Welcome XianYi Lian to join the Gao lab. Congratulation her on winning the Peking University Boya Postdoctoral Fellowship!
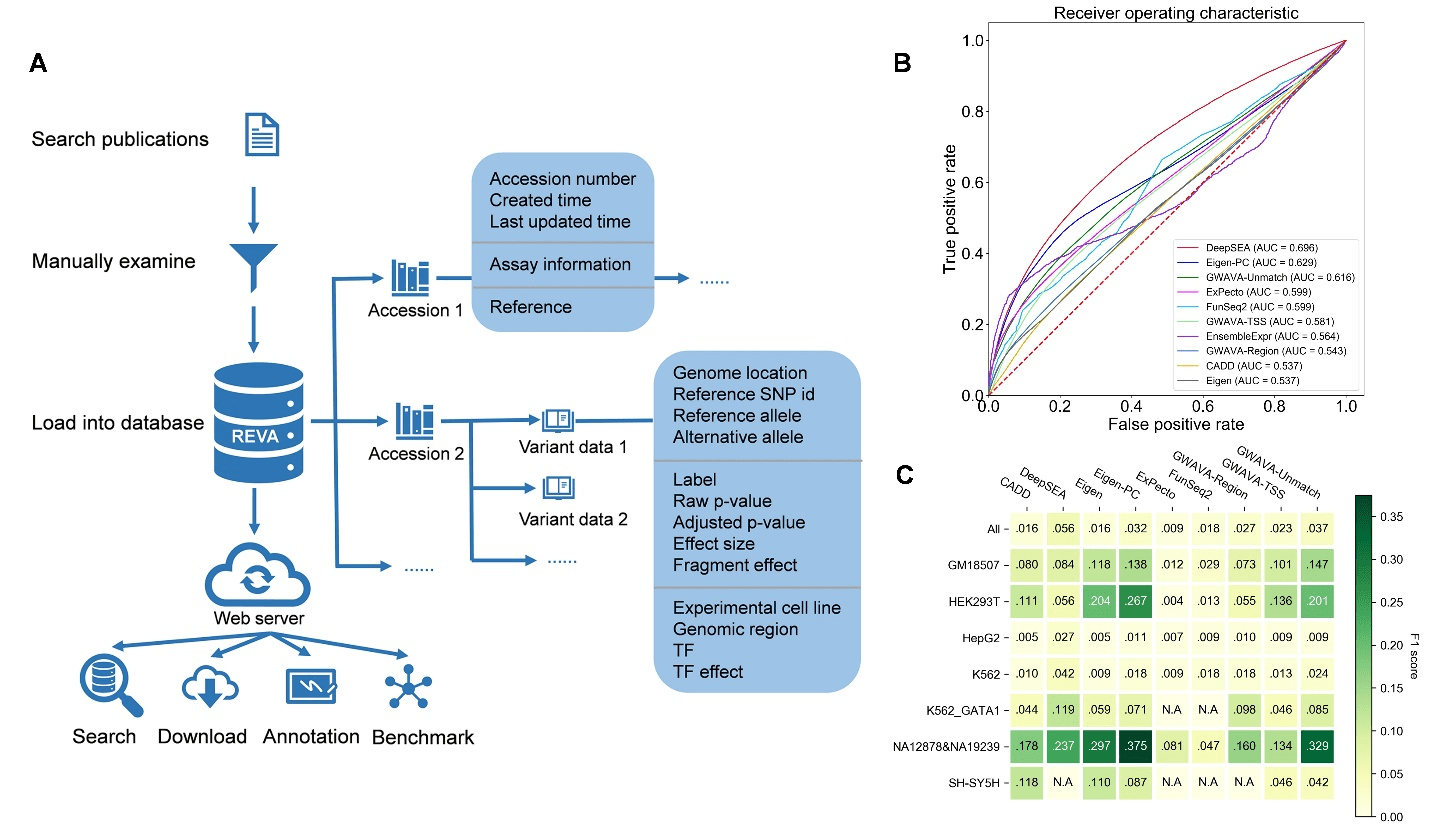
REVA: Systematical curation and evalutation for human expression-modulating variants
Approximately 97% of the human genome is noncoding. While more than 90% of disease- and trait-associated variants are noncoding variants, their biological functions and mechanisms remain largely elusive. Recently, Gao Lab from Biomedical Pioneering Innovation Center (BIOPIC), Beijing Advanced Innovation Center for Genomics (ICG), Center for Bioinformatics (CBI), and State Key Laboratory of Protein Read more about REVA: Systematical curation and evalutation for human expression-modulating variants[…]

Congratulations to Fangyuan Shi for her Ph.D.!
Well done, Dr. Shi! Wish you more success and prosperity in days to come!