Approximately 97% of the human genome is noncoding. While more than 90% of disease- and trait-associated variants are noncoding variants, their biological functions and mechanisms remain largely elusive.
Recently, Gao Lab from Biomedical Pioneering Innovation Center (BIOPIC), Beijing Advanced Innovation Center for Genomics (ICG), Center for Bioinformatics (CBI), and State Key Laboratory of Protein and Plant Gene Research at School of Life Sciences, compiled REVA as the largest database for experimentally tested human expression-modulating noncoding variants by systematically curating multiple published studies, and further benchmarked seven popular computational tools based on high-quality data in REVA.
To ensure unified and high-quality data, all records in REVA were collected and curated using a well-defined protocol. The current release of REVA consists of over 11.8 million expression-modulating variants experimentally tested in 18 distinct cell lines. In efforts to pinpoint plausible regulatory mechanisms for these variants, they annotated the functional effects of these variants based on 2,403 trained convolutional neural networks (CNNs) covering transcription factors binding and, epigenetic modifying events, as well as open chromatin regions. Moreover, the annotation page also incorporated 21 DNA physicochemical properties and evolutionary features.
While multiple computational tools have been developed for identifying expression-modulating variants, there is no comprehensive evaluation of these computational tools based on high-quality expression-modulating variants. The authors benchmarked seven state-of-the-art computational tools based on high-quality data in REVA and found limited sensitivity of current tools remains a serious challenge for effective large-scale analysis. Of note, all benchmarked tools showed different performance on different types of variants (cell lines/conservation/traits associated/diseases associated), further demonstrating the complex nature of gene regulation machinery for noncoding variants.
REVA (http://reva.gao-lab.org) is available as an interactive web server for users to explore all data entries and analysis results. Users can launch a quick search by chromosome position, rs id, gene name, ensembl gene id, or disease name. “Advanced search” provides a customized search and batch search for users. Abundant annotation is available for each queried variant online through eight modules: “Basic information”, “Cell Line and Expression”, “Three-dimensional Interacting Gene”, “Chromatin State”, “Disease and Phenotype”, “Meta Sources”, “Accession”, and “Annotation”. Users can also download the annotation for further analysis and check the benchmarking results of state-of-the-art computational tools.
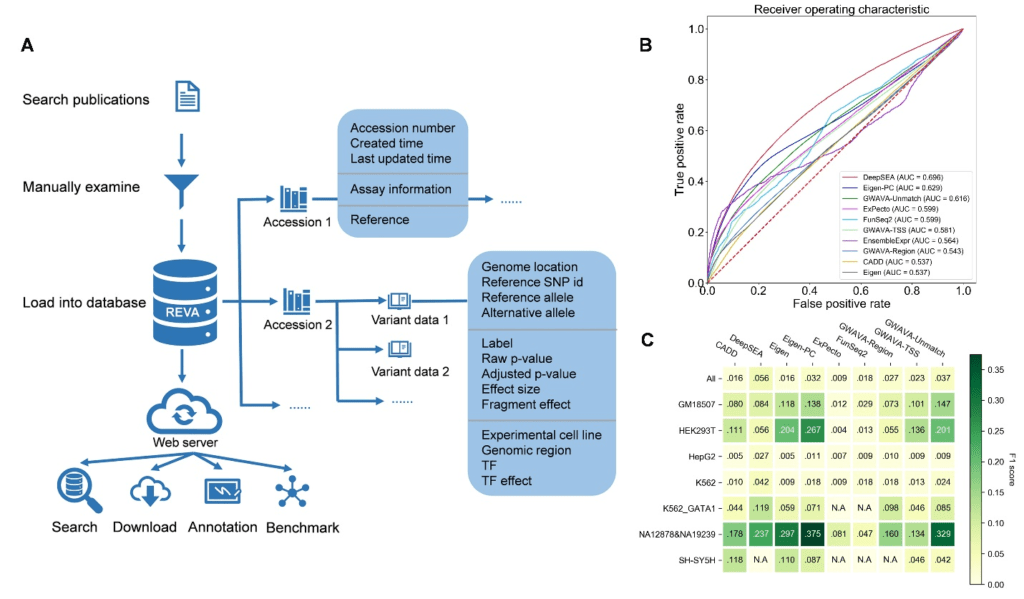
Figure 1:Overview of the structure of REVA and part of evaluation results. A. Overview of the structure of REVA. B. Performance of involved tools on the benchmarking dataset. C. Performance comparison of involved tools except EnsembleExpr on variants from different cell lines.
The work has been published online in Genomics, Proteomics & Bioinformatics. Yu Wang and Fang-Yuan Shi from School of Life Sciences, Peking University are co-first authors and Dr. Ge Gao is the corresponding author. Yu Liang from School of Life Sciences, Nanchang University made contribution to the data collection. This work was supported by funds from the National Key Research and Development Program of China and the National High Technology Research and Development Program of China as well as the State Key Laboratory of Protein and Plant Gene Research and the Beijing Advanced Innovation Center for Genomics (ICG) at Peking University. The research of Ge Gao was supported in part by the National Program for Support of Top-notch Young Professionals. Part of the analysis was performed on the Computing Platform of the Center for Life Sciences of Peking University and was supported by the High-performance Computing Platform of Peking University.
Link:
https://www.sciencedirect.com/science/article/pii/S167202292100142X