scRNA-seq has seen rapid development over the recent years, providing powerful technological support for studying important biological problems including cellular functions and gene regulatory networks. In a typical scRNA-seq study, cell annotation regarding cell types and cell differentiation progress is usually the first step. However, the standard annotation process is rather tedious, and cannot guarantee comparability across different studies. As scRNA-seq data continues to accumulate, utilizing existing data as references to annotate newly sequenced cells is becoming a potential solution to the problem.
Utilizing existing data involves comparison across different scRNA-seq datasets, which is often complicated by “batch effect”. Batch effect is typically a cocktail of numerous types of cross-dataset differences, including but not limited to the difference in transcript capture rate and sequence bias of various scRNA-seq protocols, operational difference among multiple experimental batches, different sequencing depths among multiple sequencing runs, different transcription regulation in different species, and even difference in bioinformatic analysis pipelines. Without effective batch effect correction, the reliability of cross-dataset comparison would be significantly undermined, impeding the utilization of existing datasets.
Generative adversarial networks (GAN) is an important development in the field of deep learning. GANs train generator networks to generate realistic samples indistinguishable with target distributions, via introducing adversarial discriminator networks to guide the generators, which has led to significant breakthroughs in various fields like image and text generation. Benefiting from its powerful distribution fitting capability, adversarial training techniques have been successfully applied to fields other than generative learning, including domain adaptation1, and has the potential to solve the problem of batch effect in single-cell genomics.
As such, we developed a scRNA-seq data querying and annotation method called Cell BLAST based on deep adversarial learning, as well as a well-curated scRNA-seq database ACA, to provide new tool and resources to facilitate utilizing existing data for reference-based cell annotation and cross-dataset studies.
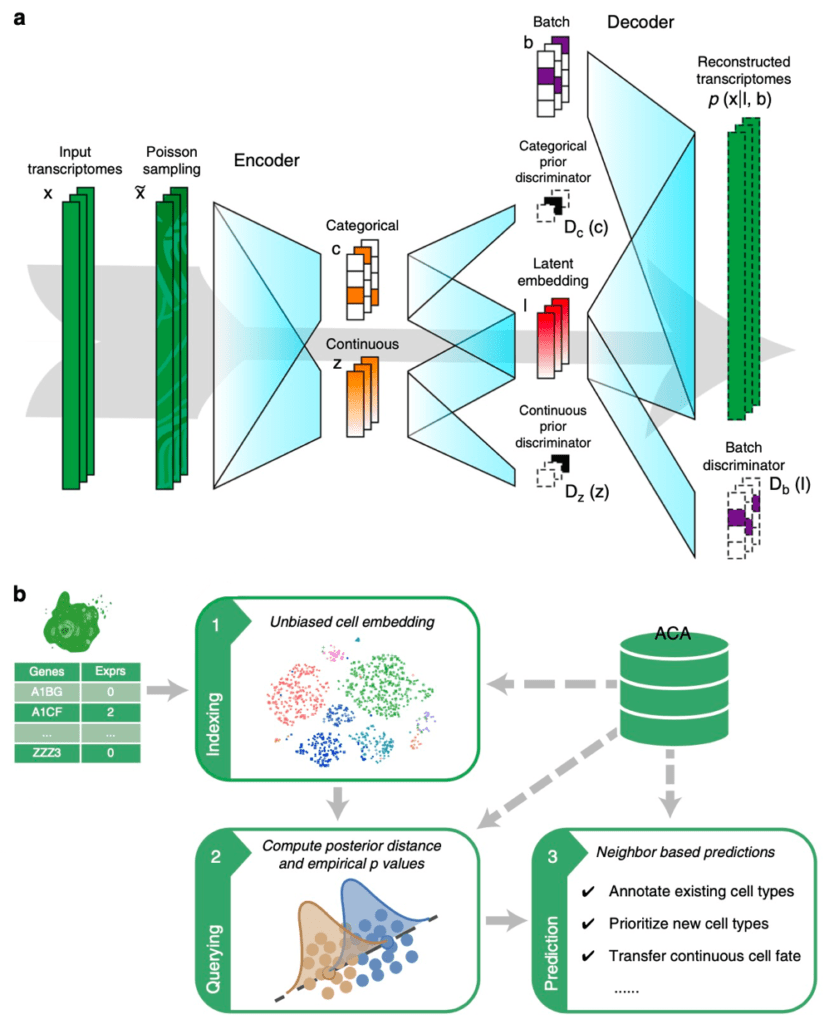
Figure 1 Cell BLAST model and workflow. (a) Structure of the generative model used by Cell BLAST. (b) Overall Cell BLAST workflow.
Analogous to sequence BLAST, Cell BLAST can retrieve cells most similar to user provided query cells in several reference datasets, and annotate the query cells based on annotation information of these database “hits”. However, batch effect between datasets will significantly affect querying reliability. To solve this problem, Cell BLAST uses adversarial autoencoder to reduce data dimensionality, and applies domain adversarial learning to eliminate batch effect between datasets (Fig. 1a). Benchmark experiments show that Cell BLAST outperforms existing batch effect correction methods (Fig. 2a). In addition, we also proposed a more accurate cell-to-cell similarity metric called NPD, based on the model posterior distribution. By design, NPD considers the intrinsic uncertainty of single-cell transcriptomics observation. Cell BLAST also gives a P-value for each query “hit” based on the empirical distribution of NPD. Benchmark experiments show that Cell BLAST has higher accuracy in cell type identification than other cell querying tools (Fig. 2b, c). Through gradient backpropagation, we further verified that the Cell BLAST model correctly captured cell type marker genes, suggesting certain level of model interpretability.
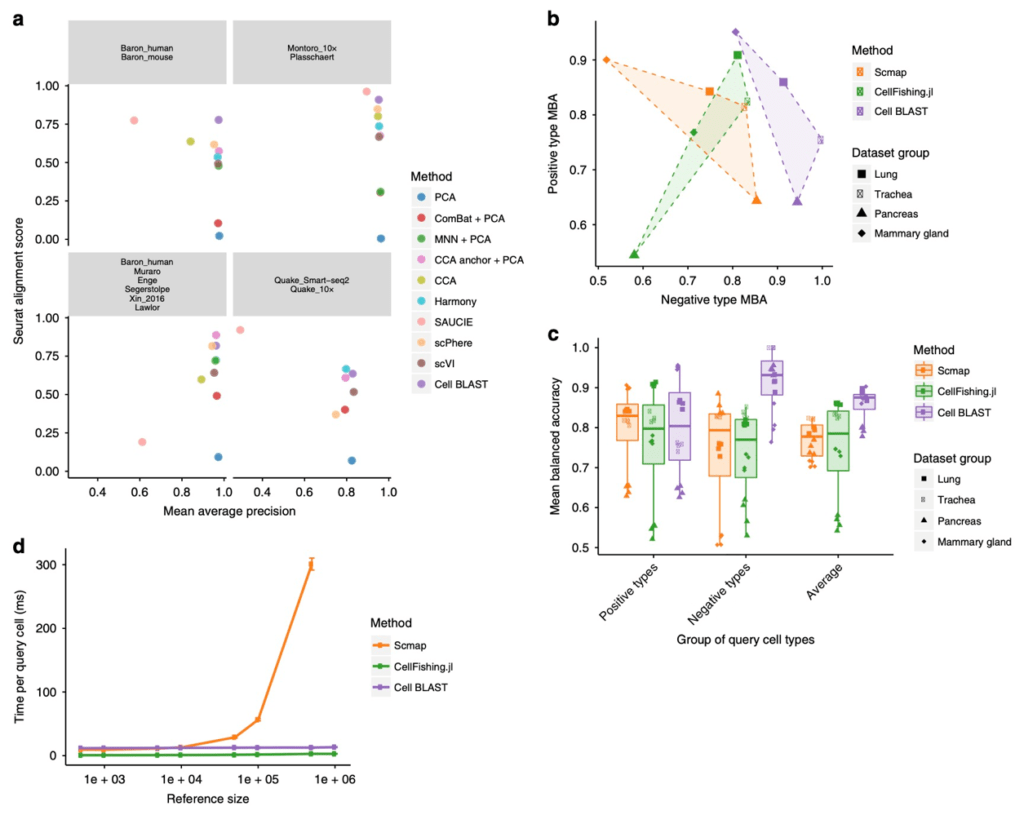
Figure 2 Cell BLAST benchmarking. (a) Comparison of batch effect correction performance. (b) Accuracy on positive and negative queries in querying-based cell typing. (c) Comparison of overall performance in querying-based cell typing. (d) Comparison of querying speed.
Apart from identifying known cell types, Cell BLAST can sensitively identify cell types non-existent in the reference data. Two back-to-back scRNA-seq studies recently published in Nature identified a rare tracheal cell type called ionocyte2, 3. Taking these studies as an example, the ionocytes in one of the datasets were artificially removed and used as a reference to annotate the other dataset. Cell BLAST sensitively discovered the existence of query ionocytes and rejected them, rather than incorrectly predicting them as other known cell types (Fig. 3a-c). In addition, we also used human and mouse hematopoietic datasets4, 5 to verify that Cell BLAST can be used in a more challenging task of annotating continuous cellular traits across different species (Fig. 3d-f). Compared with other existing tools, cell differentiation fate predicted by Cell BLAST exhibits a higher correlation with the expression level of known fate-determining genes.
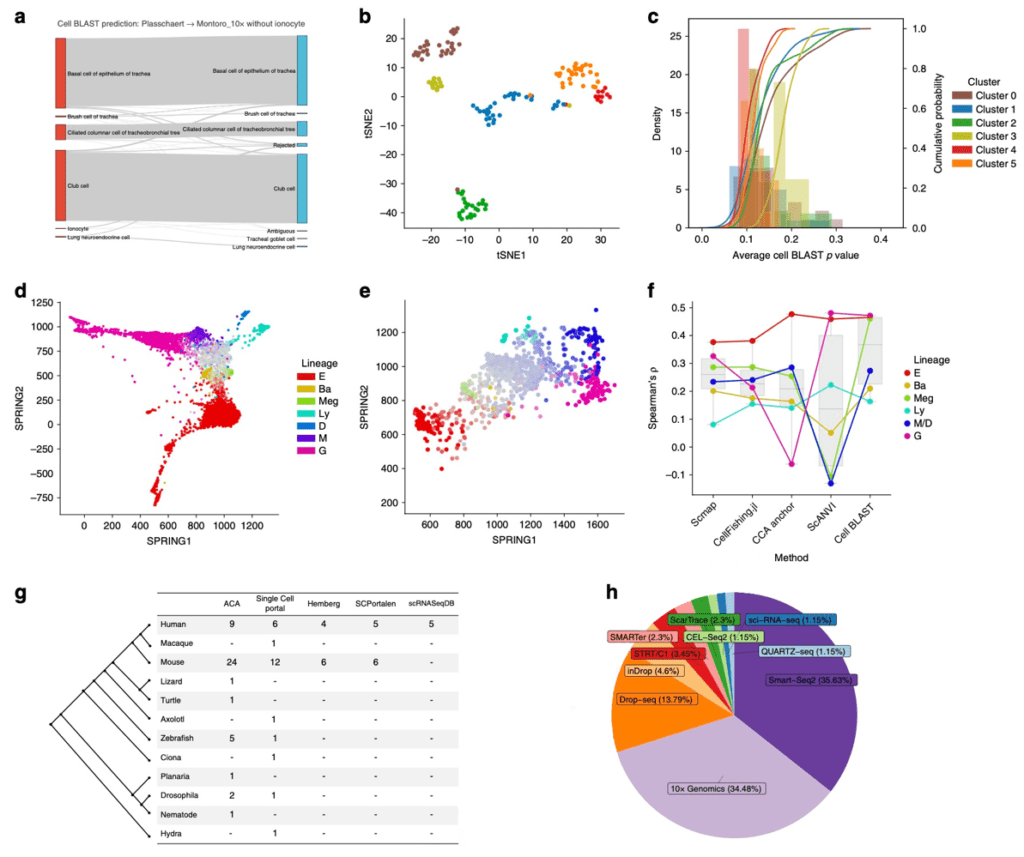
Figure 3 Cell BLAST application. (a) Prediction result of using the ionocyte-removed reference tracheal dataset to annotate another complete tracheal dataset. (b) Clustering of rejected cells in the above querying result. (c) P-value distribution of the different rejected clusters, in which the cluster with largest P-value (least similar to reference cells) is composed of ionocytes. (d) Visualization of the mouse hematopoietic differentiation dataset (colors indicate terminal fates, while saturation indicate differentiation progress). (e) Using the mouse hematopoietic dataset to annotate the cell fate of a human dataset. (f) Comparison of cross-species cell fate annotation preformance using different computational methods. (g) Comparison of dataset number in ACA and other scRNA-seq databases. (h) Dataset proportion of different scRNA-seq protocols in ACA.
Just like sequence BLAST, pratical application of Cell BLAST requires a large-scale well-curated database. By collecting a large number of published scRNA-seq datasets, we established a database covering 2,989,582 single cells across 8 species and 27 different organs, which we termed the Animal Cell Atlas (ACA) (Fig. 3g, h). We extensively organized the cell annotations in ACA and used Cell Ontology to construct a set of structured cell type annotations, to unify the annotations in different datasets and support ontology-aware inference of cell types.
Cell BLAST provides an online cell querying platform (https://cblast.gao-lab.org). Users can directly upload their scRNA-seq data to perform cell querying and automatic annotation based on the reference datasets in ACA. Meanwhile, an open source Python package Cell BLAST (https://github.com/gao-lab/Cell_BLAST) is also provided, which enables model training, cell querying and customized analysis on custom reference datasets.
Citation:
Websites:
- Web Server: https://cblast.gao-lab.org
- Source Code: https://github.com/gao-lab/Cell_BLAST
References:
- Tzeng E, Hoffman J, Saenko K, Darrell T. Adversarial discriminative domain adaptation. In IEEE Conference on Computer Vision and Pattern Recognition. 7167-7176 (2017).
- Montoro DT, et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319-324 (2018).
- Plasschaert LW, et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377-381 (2018).
- Tusi BK, et al. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 555, 54-60 (2018).
- Velten L, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol 19, 271-281 (2017).