Life is an organized arrangement of cells, but any individual cell struggles to function independently once detached from a living organism. Therefore, a comprehensive understanding of cell functionality requires considering the microenvironment and spatial location in which cells exist. The rapid development of spatial omics technology greatly facilitates the study of cell function at the spatial and single-cell precision, as well as the multi-cellular regulatory processes within tissues. Specifically, aligning and comparing different spatial omics slices contributes to revealing differences in the spatial distribution of cells during processes such as changes over time and in diseases, then deciphering the underlying biological mechanisms. However, current mainstream computational methods mainly focus on homogeneous slices (such as spatial domain segmentation of the same slice and reconstruction of multiple slices from the same tissue), making it challenging to handle heterogeneous slices across time and space, and modalities.

In order to address these challenges, on November 9, 2023, Dr. Ge Gao’s lab at Peking University/Changping Laboratory published a research paper in Nature Communication titled “Spatial-linked alignment tool (SLAT) for aligning heterogeneous slices.”, introducing the SLAT method, which is a cross-platform, multi-modal spatial omics alignment, and integration approach. SLAT can be applied to many scenarios such as cross-omics spatial alignment and continuous spatiotemporal developmental analysis.
To effectively handle heterogeneous slices, SLAT models spatial data as a graph, considering cells as nodes and edges between nodes representing the proximity relationships between cells. It utilizes graph neural networks to aggregate cellular neighborhood information, obtaining spatial cell representations. Through adversarial learning, SLAT further eliminates topological heterogeneity among graphs, ensuring that cell representations from different graphs locate in the same manifold, achieving precise alignment across platforms and multi-omics heterogeneous slices (Figure 1).
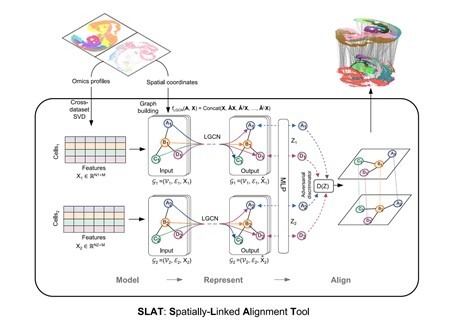
Figure 1. Architecture of the SLAT model
One challenge in spatial omics data alignment arises from significant differences in resolution among various spatial omics technology platforms. For instance, 10x Visium has a resolution of 50 μm, Spatial ATAC-seq has a resolution of 20 μm, and Stereo-seq has a resolution of 0.2 μm, reaching differences of two orders of magnitude. SLAT introduces the concept of variable neighborhoods, employing different neighborhood sizes during graph construction for technologies with distinct resolutions. This ensures the spatial representations of cells from heterogeneous spatial data are comparable, resulting in excellent alignment across platforms (Xenium and Visium, Stereo-seq and seqFISH) and cross-omics (Stereo-seq and spatial-ATAC-seq), as depicted in Figure 2.
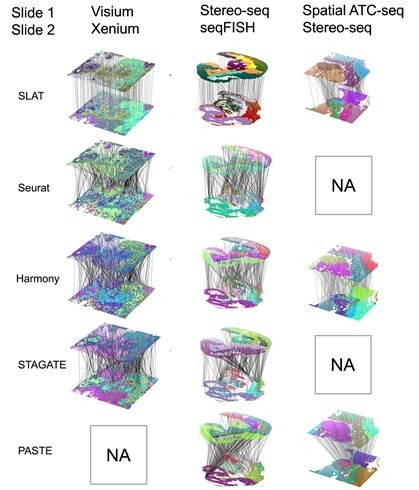
Figure 2. SLAT outperforms other methods in aligning heterogeneous data across platforms and omics
Based on these technological innovations, SLAT enables precise tracking of complex spatiotemporal changes, such as in early embryonic development. Analyzing consecutive spatiotemporal slices of mouse development from E9.5 to E16.5 (Figure 3a), SLAT accurately reconstructs the large-scale developmental process from intermediate kidney (E11.5) to mature kidney (E12.5) (Figure 3b).
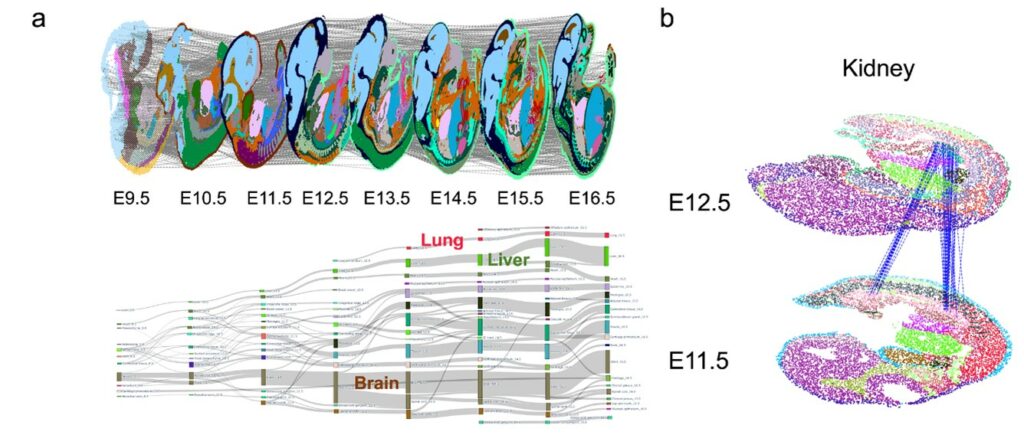
Figure 3. SLAT can accurately align spatiotemporal slice data at different developmental stages
The code of SLAT is publicly available at https://github.com/gao-lab/SLAT. I Users can install and use the software package directly through PyPI or Anaconda.
Ph.D. candidate Chen-Rui Xia from Peking University’s School of Life Sciences is the first author of the paper. Undergraduate student Xin-Ming Tu (currently a Ph.D. candidate in the Department of Computer Science and Engineering at the University of Washington) is a co-author. Dr. Zhi-Jie Cao is the corresponding co-author and co-first author of the paper. This research received support from the National Key Research and Development Program, the State Key Laboratory of Protein and Plant Gene Research, the Beijing Advanced Innovation Center for Genomics, and Changping Laboratory. Computational analysis was conducted on the high-performance computing platforms at Changping Laboratory, Pacific High-Performance Computing Platform of Peking University, and High-performance Computing Platform of Peking University.
Link to the paper: https://doi.org/10.1038/s41467-023-43105-5