Nat. Biotechnol. | An accurate and robust computational method for single-cell multi-omics integration and regulatory inference
Gene transcription is a key link in the central dogma of biology. Compared to the relative static genome, the transcriptome exhibits substantial changes across different tissues, organs and developmental stages, forming crucial biological basis for the physiological and pathological function of cells. Cells are the basic units of life. Rapid advances in single-cell sequencing technologies provide valuable tools for investigating cellular functions and their underlying gene regulatory mechanisms at single-cell resolution. A number of omics modalities can be probed by single-cell sequencing, including transcriptome, chromatin accessibility, DNA methylation, histone modification, etc. Integrative analysis of data in different omics modalities promises more holistic characterization of cellular states and regulatory circuits. However, compared with conventional bulk omics data, single-cell data features large data size (up to millions of cells), high level of noise (dropout, batch effect), as well as high heterogeneity. Developing novel computational methods to utilize these valuable data effectively has become a focus and hotspot in the field of bioinformatics.
In order to address these challenges, on May 2, 2022, Dr. Ge Gao’s lab at Peking University / Changping Laboratory, published a research article in Nature Biotechnology titled “Multi-omics single-cell data integration and regulatory inference with graph-linked embedding”, releasing a deep learning model called GLUE based on a proposed graph-linking strategy, which, for the first time, achieved accurate unsupervised multi-omics integration and regulatory inference at the scale of millions of cells.

The key challenge of computational multi-omics integration is the discrepancy in feature space among different omics layers. For example, the feature space of transcriptome consists of genes, while that of chromatin accessibility consists of open chromatin regions. Such discrepancy causes a lack of comparability between cells in different omics layers. To address this problem, the authors proposed a novel graph-linking strategy. Specifically, prior knowledge regarding regulatory interactions between omics features is encoded into a guidance graph, in which nodes are omics features, and edges represent prior regulatory knowledge. The GLUE model then employs a variational graph autoencoder (VGAE) to learn low-dimensional feature embeddings from the guide graph, which serves as the weights of linear data decoders, effectively linking different autoencoders to ensure “semantic consistency” of the learned cell embeddings. Finally, adversarial learning between the encoders and a modality discriminator was applied to ensure alignment (Fig. 1).
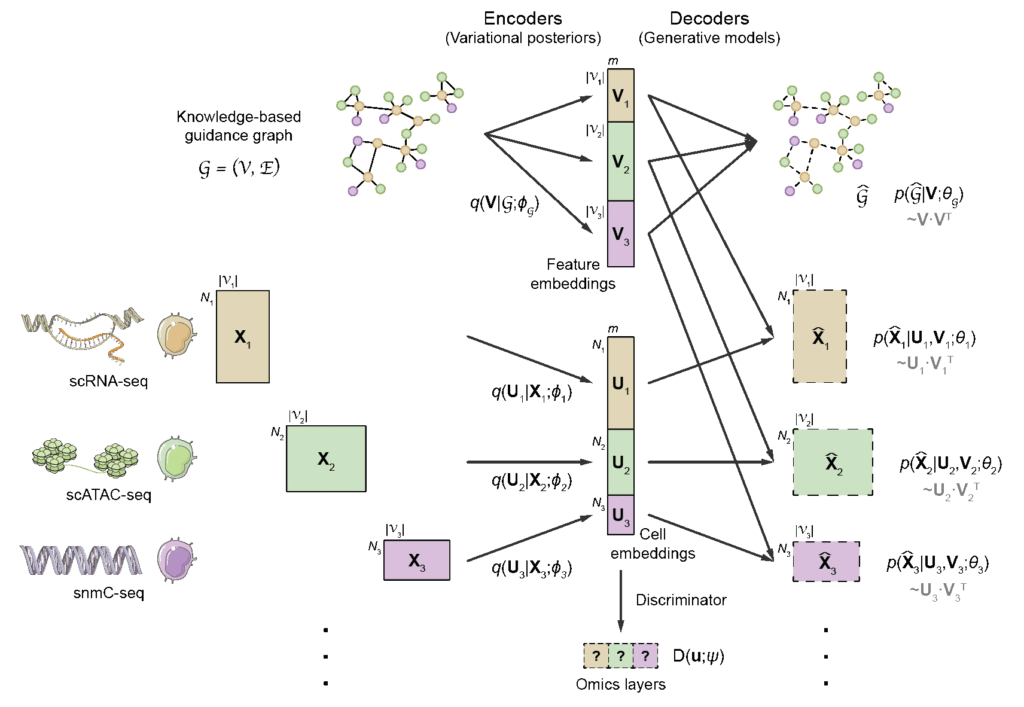
Fig. 1 Architecture of the GLUE model.
Main advantages of the GLUE model include:
- High accuracy in multi-omics integration: Systematic evaluation using multiple single-cell transcriptomics and chromatin accessibility datasets demonstrated that GLUE exhibits higher integration accuracy than state-of-the-art algorithms, at both cell type and single-cell levels ( 2a–c).
- High robustness to prior regulatory knowledge: The prior regulatory knowledge used in the GLUE guidance graph does not need to be precise. Taking the integration of transcriptomics and chromatin accessibility data as an example, sketchy connections between open chromatin regions and adjacent genes is sufficient for effective integration. Corruption experiments showed that GLUE remains accurate even with significantly corrupted guidance graphs ( 2d).
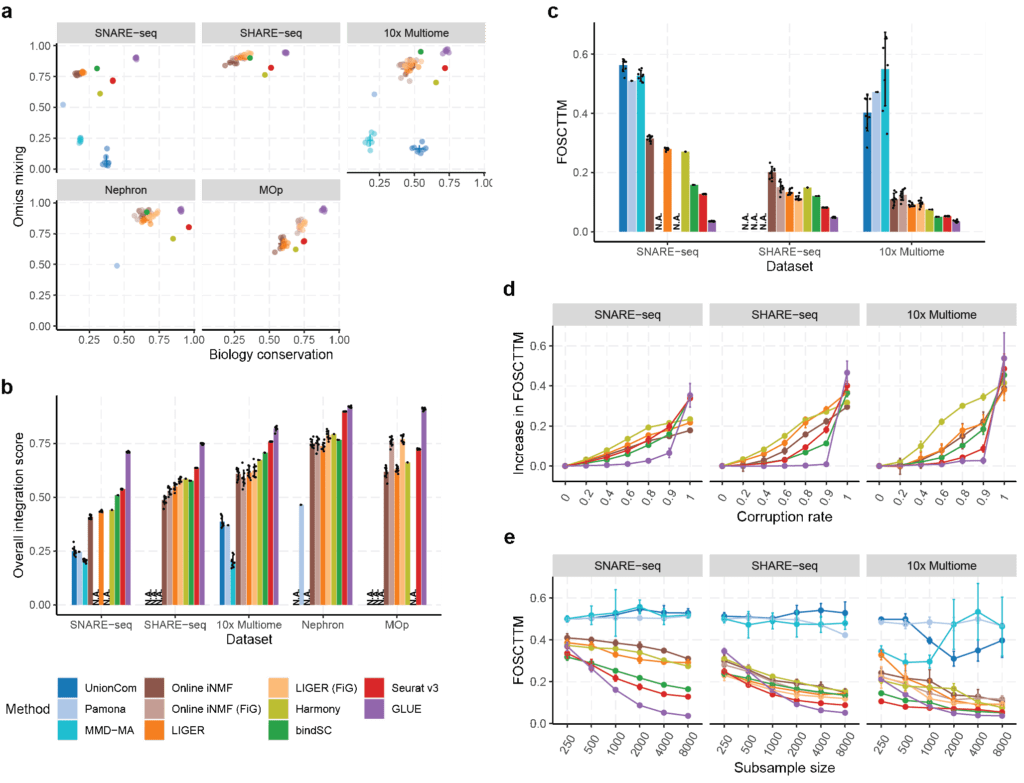
Fig. 2 Systematic evaluations of multi-omics integration accuracy.
- High scalability to big data: GLUE achieves sublinear computational scalability, making it the first method to achieve accurate multi-omics integration over millions of cells ( 3).

Fig. 3 GLUE scales to atlas-scale integration containing millions of cells.
- Supporting the integration of arbitrary numbers of omics layers with arbitrary regulatory directions: By stacking multiple omics-specific variational autoencoders (VAEs), GLUE supports unsupervised integration of unpaired omics modalities. The authors successfully applied GLUE to a triple omics integration (transcriptome, chromatin accessibility, and DNA methylation) of the mouse cortex, and showed that the integration can further improve cell type annotation. Meanwhile, GLUE is modular by design, and can be readily extended to support additional modalities like single-cell Ribo-seq, spatial transcriptomics, etc.
- Capability of integrative regulatory inference: Apart from cell-level integration, GLUE explicitly models regulatory interactions in the form of a guidance graph, enabling further integration of prior regulatory knowledge and observed correlation in the integrated multi-omics data to achieve reliable regulatory inference. Using PBMC (peripheral blood mononuclear cells) data as an example, the authors applied GLUE to integrate physical interactions from pcHi-C, genetic associations from eQTL, as well as single-cell transcriptomics and chromatin accessibility data. The results demonstrated that by combining multiple types of regulatory evidence, GLUE yields more reliable regulatory inference than would be possible from individual types of evidence ( 4). Again, it is worth noting that prior regulatory interactions in the guidance graph does not need to be precise. Systematical evaluations revealed that GLUE-based multi-omics integration and regulatory inference both demonstrate significant robustness.
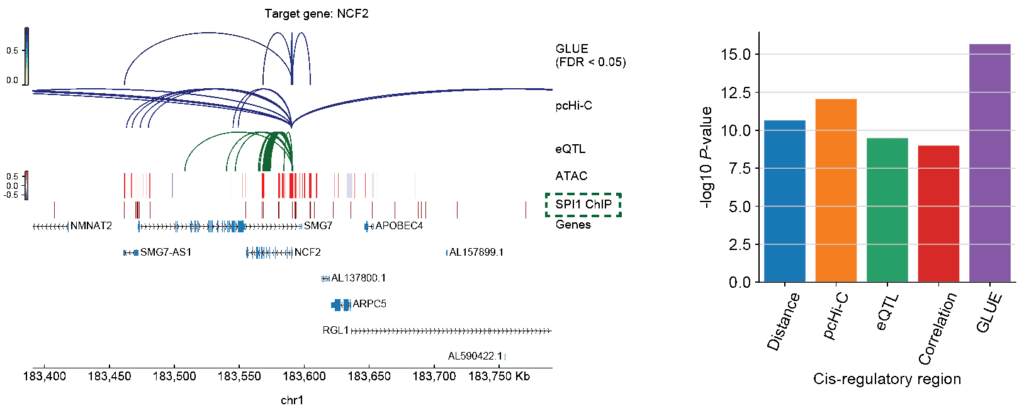
Fig. 4 GLUE is capable of integrative regulatory inference combining both prior regulatory knowledge and observed single-cell multi-omics data.
The code of GLUE is publicly available at https://github.com/gao-lab/GLUE. Users can install and use the software package directly through PyPI or Anaconda.
PhD student Zhi-Jie Cao is the first author of the study. Professor Ge Gao is the corresponding author. The study was supported by funds from the National Key Research and Development Program, the State Key Laboratory of Protein and Plant Gene Research, the Beijing Advanced Innovation Center for Genomics at Peking University, as well as the Changping Laboratory. Part of the analysis was carried out on the Computing Platform of the Center for Life Sciences of Peking University and supported by the High-performance Computing Platform of Peking University.