Gene transcription and protein translation are two key steps of the “central dogma”. Cells often response to disease and environment stress via transcriptional and translational control. Meanwhile, besides coding proteins, RNAs could function as noncoding molecules. Benefitting from bioinformatics and computational genomics methods, it is possible to quantitatively de-convolute factors contributing to translational control in cellular regulatory map via modelling coding ability of transcripts.
Recently, Gao Lab from Biomedical Pioneering Innovation Center (BIOPIC), Beijing Advanced Innovation Center for Genomics (ICG), Center for Bioinformatics (CBI), and State Key Laboratory of Protein and Plant Gene Research at School of Life Sciences, developed RiboCalc (Ribosome Calculator) as an experiment-backed, data-oriented computational model for quantitatively predicting the coding ability (Ribo-seq expression level) of a particular human transcript. The work has been published online in Briefings in Bioinformatics.

Gao Lab collected reliable paired Ribo-seq/RNA-seq data from the GEO database, covering 22 different cell types. By applying rigorous filtering criteria, the translation status for 101,170 GENCODE transcripts were called, among which, the translation status of 46% were found to be “coding” while 43% were “noncoding” in all samples. Interestingly, there were 11% of the transcripts exhibited diverse coding ability among cell lines (i.e., coding in some cell lines but noncoding in others), and named as context-dependent coding transcripts (CDCTs).
With data-driven feature selection and model building strategy, Ribo was built to quantitatively predict transcripts’ coding ability (TPM in Ribo-seq) across cell lines with (intrinsic, cis-) and (contextual, trans-) features,showing high accuracy (r = 0.81). The model implies that both transcript sequence and cell environment play important roles in coding ability determination, suggesting transcript’s coding ability should be modeled as a context-dependent continuous value, rather than a certain binary class. Remarkably, ncRNAs were reported to be associated with ribosomes and even encoded peptides since 2014 [1-2]. Predicted by RiboCalc, the ribosome-associated ncRNAs were with higher coding score, providing novel hint for this “coding ncRNA” paradox.
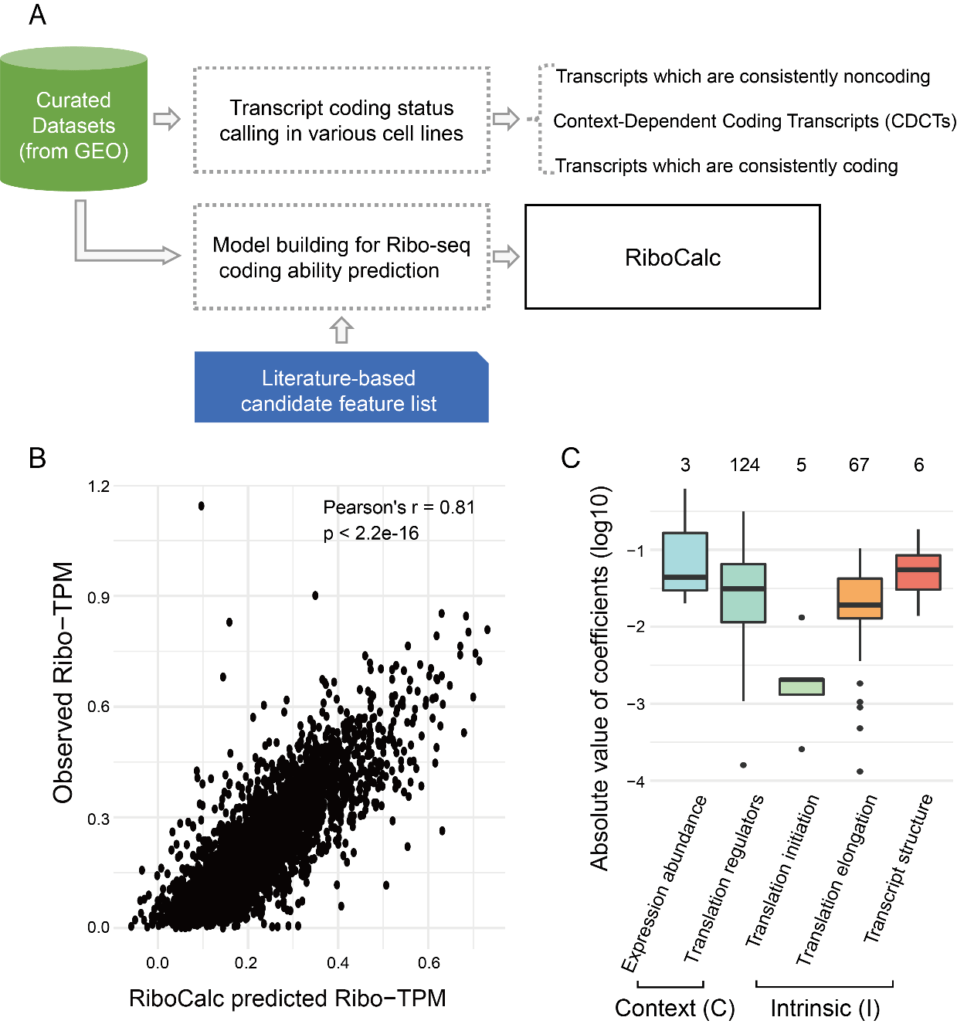
Figure 1, workflow and main results of RiboCalc. A, workflow of RiboCalc model building. B, performance of RiboCalc in independent testing. C, contribution of each class of features in the model.
As the first attempt in mammalians, RiboCalc accurately predict coding ability across cell lines in human. The program and tutorial are published at https://github.com/gao-lab/RiboCalc/.
Feel free to mail [email protected] if you have any issues!
Yu-Jian Kang, the postdoc from School of Life Sciences, Peking University is the first authors and Dr. Ge Gao is the corresponding author. This work received great support from Jing-Yi Li, Lan Ke, Shuai Jiang and De-Chang Yang from School of Life Sciences, Peking University. This work was supported by funds from the National Key Research and Development Program of China and the National High Technology Research and Development Program of China as well as the State Key Laboratory of Protein and Plant Gene Research and the Beijing Advanced Innovation Center for Genomics (ICG) at Peking University. The research of Ge Gao was supported in part by the National Program for Support of Top-notch Young Professionals. Part of the analysis was performed on the Computing Platform of the Center for Life Sciences of Peking University and was supported by the High-performance Computing Platform of Peking University.
Link:
References:
- Ruiz-Orera J, Messeguer X, Subirana JA et al. Long non-coding RNAs as a source of new peptides, Elife 2014;3:e03523.
- Zeng C, Fukunaga T, Hamada M. Identification and analysis of ribosome-associated lncRNAs using ribosome profiling data, BMC Genomics 2018;19:414.